

Fitting the pair potentials for molten salts: A review in brief
Abstract
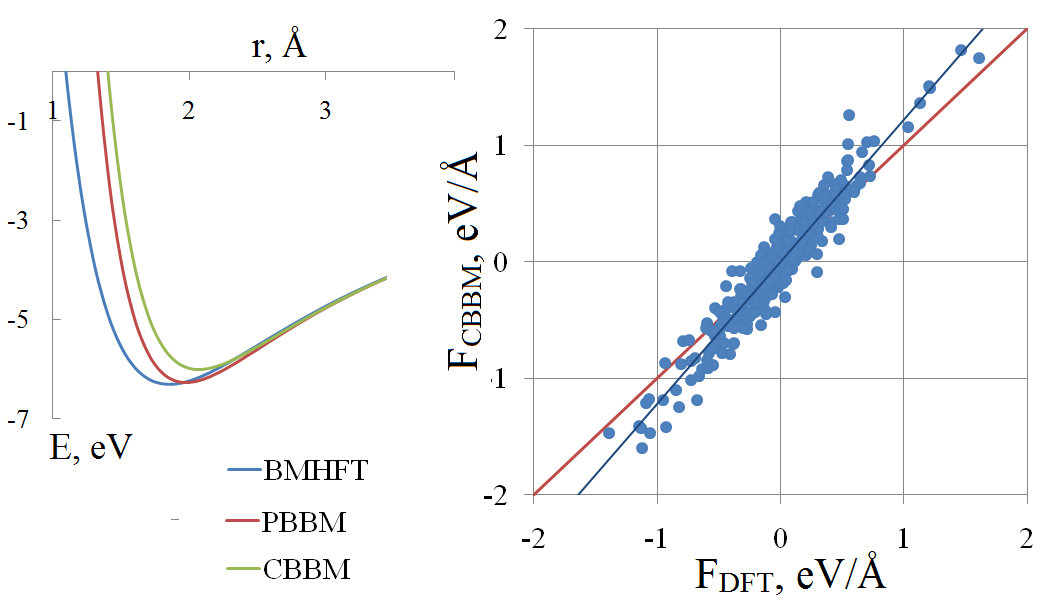
In vitro and in silico studies should supplement each other in order to obtain reliable and comprehensive data on physicochemical properties of molten salts. To attain the aim, the appropriate simulation technique is needed. Because of the computational speed that classical molecular dynamics could deliver, this method is often the most suitable for calculation of the transport properties. The accuracy of calculation is to a high degree depending on parameters of the potential. In this paper, we review the basics of the pair potential fitting procedure. As an example, a molten lithium chloride is considered. The comparison of different pair potentials in terms of potential energy and per-atomic forces is performed, with the reference data were obtained by means of the density functional theory. Among the macroscopic properties, the melting temperature and viscosity are calculated.
Keywords
Full Text:
PDFReferences
Pople JA, Nobel Lecture: Quantum chemical models, Rev. Mod. Phys. 71 (1999) 1267–1274. https://doi.org/10.1103/revmodphys.71.1267
Geerlings P, De Proft F, Langenaeker W, Conceptual Density Functional Theory, Chem. Rev. 103 (2003) 1793–1874. https://doi.org/10.1021/cr990029p
Hohenberg, P, Kohn W, Inhomogeneous Electron Gas, Phys. Rev. 136 (1964) B864–71. https://doi.org/10.1103/physrev.136.b864
Kohn W, Sham LJ, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev. 140 (1965) A1133–A1138. https://doi.org/10.1103/physrev.140.a1133
Cohen AJ, Mori-Sánchez P, Yang W, Challenges for Density Functional Theory, Chem. Rev. 112 (2011) 289–320. https://doi.org/10.1021/cr200107z
Fedik N, Zubatyuk R, Kulichenko M, Lubbers N, et al., Extending machine learning beyond interatomic potentials for predicting molecular properties, Nat. Rev. Chem. 6 (2022) 653–672. https://doi.org/10.1038/s41570-022-00416-3
Behler J, Parrinello M, Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces, Phys. Rev. Lett. 98 (2007) 146401. https://doi.org/10.1103/physrevlett.98.146401
Behler J, Constructing high-dimensional neural network potentials: A tutorial review, Int. J. Quantum Chem. 115 (2015) 1032–1050. https://doi.org/10.1002/qua.24890
Tissen JTWM., Janssen GJM., Molecular-dynamics simulation of molten alkali carbonates, Mol. Phys. 71 (1990) 413–426. https://doi.org/10.1080/00268979000101871
Ding J, Du L, Pan G, Lu J, et al., Molecular dynamics simulations of the local structures and thermodynamic properties on molten alkali carbonate K2CO3, Appl. Energy. 220 (2018) 536–544. https://doi.org/10.1016/j.apenergy.2018.03.116
Du L, Xie W, Ding J, Lu J, et al., Molecular dynamics simulations of the thermodynamic properties and local structures on molten alkali carbonate Na2CO3, Int. J. Heat Mass Transf. 131 (2019) 41–51. https://doi.org/10.1016/j.ijheatmasstransfer.2018.11.044
Seo W-G, Matsuura H, Tsukihashi F, Calculation of phase diagrams for the FeCl2, PbCl2, and ZnCl2 binary systems by using molecular dynamics simulation, Metall. Mater. Trans. B 37 (2006) 239–251. https://doi.org/10.1007/bf02693154
Lee S-C, Zhai Y, Li Z, Walter NP, et al., Comparative Studies of the Structural and Transport Properties of Molten Salt FLiNaK Using the Machine-Learned Neural Network and Reparametrized Classical Forcefields, J. Phys. Chem. B. 125 (2021) 10562–10570. https://doi.org/10.1021/acs.jpcb.1c05608
Galamba N, Nieto de Castro CA, Ely JF, Molecular Dynamics Simulation of the Shear Viscosity of Molten Alkali Halides, J. Phys. Chem. B. 108 (2004) 3658–3662. https://doi.org/10.1021/jp036234x
Galamba N, Nieto de Castro CA, Ely JF, Thermal conductivity of molten alkali halides from equilibrium molecular dynamics simulations, Chem. Phys. 120 (2004) 8676–8682. https://doi.org/10.1063/1.1691735
Galamba N, Nieto de Castro CA, Ely JF, Shear viscosity of molten alkali halides from equilibrium and nonequilibrium molecular-dynamics simulations, Chem. Phys. 122 (2005) 224501. https://doi.org/10.1063/1.1924706
Wang J, Sun Z, Lu G, Yu J, Molecular Dynamics Simulations of the Local Structures and Transport Coefficients of Molten Alkali Chlorides, J. Phys. Chem. B. 118 (2014) 10196–10206. https://doi.org/10.1021/jp5050332
Zhang S, Jin Y, Yan Y, Depression of melting point and latent heat of molten salts as inorganic phase change material: Size effect and mechanism, J. Mol. Liq. 346 (2022) 117058. https://doi.org/10.1016/j.molliq.2021.117058
Wu J, Ni H, Liang W, Lu G, et al., Molecular dynamics simulation on local structure and thermodynamic properties of molten ternary chlorides systems for thermal energy storage, Comput. Mater. Sci. 170 (2019) 109051. https://doi.org/10.1016/j.commatsci.2019.05.049
Lv X, Dong A, Dai Y, Wang J et al., Temperature and concentration dependence of the physical properties and local structures of molten NaCl-KCl-LiCl mixtures, J. Mol. Liq. 229 (2017) 330–338. https://doi.org/10.1016/j.molliq.2016.12.091
Salanne M, Madden PA, Polarization effects in ionic solids and melts, Mol. Phys. 109 (2011) 2299–2315. https://doi.org/10.1080/00268976.2011.617523
Salanne M, Rotenberg B, Jahn S, Vuilleumier R, et al., Including many-body effects in models for ionic liquids, Theor. Chem. Acc. 131 (2012). https://doi.org/10.1007/s00214-012-1143-9
Wang H, DeFever RS, Zhang Y, Wu F, et al., Comparison of fixed charge and polarizable models for predicting the structural, thermodynamic, and transport properties of molten alkali chlorides, Chem. Phys. 153 (2020) 214502. https://doi.org/10.1063/5.0023225
Takagi R, Hutchinson F, Madden PA, Adya AK, et al., The structure of molten and simulated with polarizable- and rigid-ion models, J. Phys. Condens. Matter 11 (1999) 645–658. https://doi.org/10.1088/0953-8984/11/3/00
Ishii Y, Kasai S, Salanne M, Ohtori N, Transport coefficients and the Stokes–Einstein relation in molten alkali halides with polarisable ion model, Mol. Phys. 113 (2015) 2442–2450. https://doi.org/10.1080/00268976.2015.1046527
DeFever RS, Wang H, Zhang Y, Maginn EJ, Melting points of alkali chlorides evaluated for a polarizable and non-polarizable model, J. Chem. Phys. 153 (2020) 011101. https://doi.org/10.1063/5.0012253
Zakiryanov D, Kobelev M, Tkachev N, Melting properties of alkali halides and the cation-anion size difference: A molecular dynamics study, Fluid Ph. Equilibria. 506 (2020) 112369. https://doi.org/10.1016/j.fluid.2019.112369
Møller C, Plesset MS, Note on an Approximation Treatment for Many-Electron Systems, Phys. Rev. 46 (1934) 618–622. https://doi.org/10.1103/physrev.46.618
Tosi MP, Fumi FG, Ionic sizes and born repulsive parameters in the NaCl-type alkali halides—II, J. Phys. Chem. Solids 25 (1964) 45–52. https://doi.org/10.1016/0022-3697(64)90160-x
Mayer JE, Dispersion and Polarizability and the van der Waals Potential in the Alkali Halides, Chem. Phys. 1 (1933) 270–279. https://doi.org/10.1063/1.1749283
Woodcock LV, Interionic pair potentials in the alkali metal halides, J. Chem. Soc. Faraday Trans. 2. 70 (1974) 1405. https://doi.org/10.1039/f29747001405
Michielsen J, Woerlee P, v.d. Graaf F, Ketelaar JAA, Pair potential for alkali metal halides with rock salt crystal structure. Molecular Dynamics Calculations on NaCl and LiI, J. Chem. Soc. Faraday Trans. 2. 71 (1975) 1730. https://doi.org/10.1039/f29757101730
Pan G-C, Ding J, Wang W, Lu J, et al., Molecular simulations of the thermal and transport properties of alkali chloride salts for high-temperature thermal energy storage, Int. J. Heat Mass Transf. 103 (2016) 417–427. https://doi.org/10.1016/j.ijheatmasstransfer.2016.07.042
Wang J, Wu J, Sun Z, Lu G, et al., Molecular dynamics study of the transport properties and local structures of molten binary systems (Li, Na)Cl, (Li, K)Cl and (Na, K)Cl, J. Mol. Liq. 209 (2015) 498–507. https://doi.org/10.1016/j.molliq.2015.06.021
Sivaraman G, Guo J, Ward L, Hoyt N, et al., Automated Development of Molten Salt Machine Learning Potentials: Application to LiCl, J. Phys. Chem. Lett. 12 (2021) 4278–4285. https://doi.org/10.1021/acs.jpclett.1c00901
Lee S-C, Zhai Y, Li Z, Walter NP, et al., Comparative Studies of the Structural and Transport Properties of Molten Salt FLiNaK Using the Machine-Learned Neural Network and Reparametrized Classical Forcefields, J. Phys. Chem. B 125 (2021) 10562–10570. https://doi.org/10.1021/acs.jpcb.1c05608
Tovey S, Narayanan Krishnamoorthy A, Sivaraman G, Guo J, et al., DFT Accurate Interatomic Potential for Molten NaCl from Machine Learning, J. Phys. Chem. C. 124 (2020) 25760–25768. https://doi.org/10.1021/acs.jpcc.0c08870
Rodriguez A, Lam S, Hu M, Thermodynamic and Transport Properties of LiF and FLiBe Molten Salts with Deep Learning Potentials, ACS Appl. Mater. Interfaces 13 (2021) 55367–55379. https://doi.org/10.1021/acsami.1c17942
Guo J, Ward L, Babuji Y, Hoyt N, et al., Composition-transferable machine learning potential for LiCl-KCl molten salts validated by high-energy x-ray diffraction, Phys. Rev. B. 106 (2022). https://doi.org/10.1103/physrevb.106.014209
Nosé S, A unified formulation of the constant temperature molecular dynamics methods, Chem. Phys. 81 (1984) 511–519. https://doi.org/10.1063/1.447334
Adamo C, Barone V, Toward reliable density functional methods without adjustable parameters: The PBE0 model, Chem. Phys. 110 (1999) 6158–6170. https://doi.org/10.1063/1.478522
Grimme S, Antony J, Ehrlich S, Krieg H, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, Chem. Phys. 132 (2010) 154104. https://doi.org/10.1063/1.3382344
Neese F, The ORCA program system, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2 (2011) 73–78. https://doi.org/10.1002/wcms.81
Martinez JA, Yilmaz DE, Liang T, Sinnott SB et al., Fitting empirical potentials: Challenges and methodologies, Curr. Opin. Solid State Mater. Sci. 17 (2013) 263–270. https://doi.org/10.1016/j.cossms.2013.09.001
Plimpton S, Fast Parallel Algorithms for Short-Range Molecular Dynamics, J. Comput. Phys. 117 (1995) 1–19. https://doi.org/10.1006/jcph.1995.1039
Perdew JP, Burke K, Ernzerhof M, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett. 77 (1996) 3865–3868. https://doi.org/10.1103/physrevlett.77.3865
Goedecker S, Teter M, Hutter J, Separable dual-space Gaussian pseudopotentials, Phys. Rev. B. 54 (1996) 1703–1710. https://doi.org/10.1103/physrevb.54.1703
Hutter J, Iannuzzi M, Schiffmann F, Van de Vondele J, cp2k: atomistic simulations of condensed matter systems, Wiley Interdiscip. Rev. Comput. Mol. Sci. 4 (2013) 15–25. https://doi.org/10.1002/wcms.1159
Aragones JL, Sanz E, Valeriani C, Vega C, Calculation of the melting point of alkali halides by means of computer simulations, J. Chem. Phys. 137 (2012) 104507. https://doi.org/10.1063/1.4745205
Janz GJ, Tomkins RPT. Physical Properties Data Compilations Relevant to Energy Storage. IV. Molten Salts: Data on Additional Single and Multi-Component Salt Systems. U S. Government printing office: Washington; 1981. P. 280.
Porter T, Vaka MM, Steenblik P, Della Corte D, Computational methods to simulate molten salt thermophysical properties. Commun. Chem 5 (2022) 69. https://doi.org/10.1038/s42004-022-00684-6
Janz GJ. Thermodynamic and Transport Properties for Molten Salts: Correlation Equations for Critically Evaluated Density, Surface Tension, Electrical Conductance, and Viscosity Data (Supplement 2). J. Phys. Chem. Ref. Data 17 (1998).
DOI: https://doi.org/10.15826/elmattech.2023.2.010
Copyright (c) 2023 Dmitry O. Zakiryanov

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
![]()
![]()
![]()
![]()