

Molecular dynamics simulation of the Ni – FLiNaK interface: adsorption layers as origin of metal passivity
Abstract
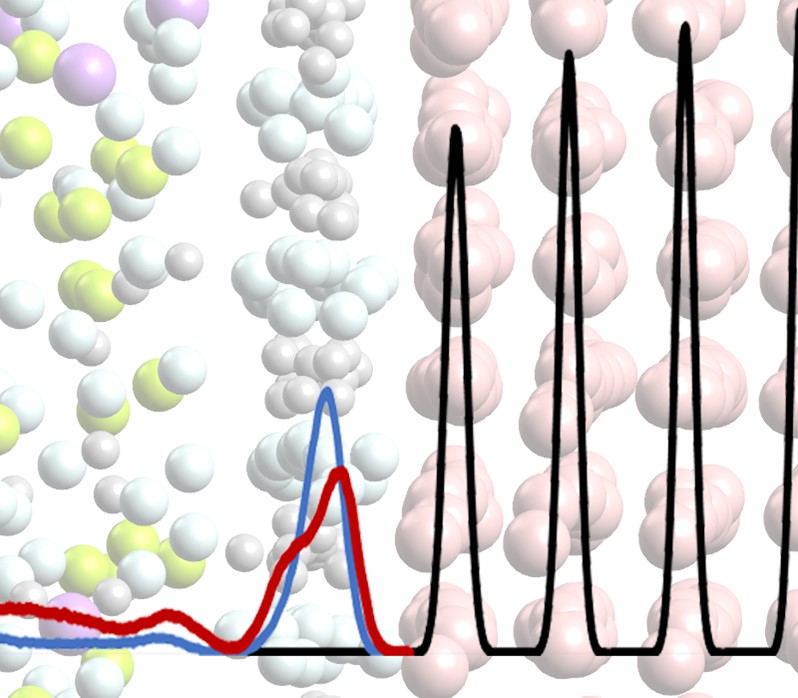
The method of classical molecular dynamics was used to study the Ni (solid) – FliNaK (melt) phase separation boundary at temperatures of 800 K, 1000K , and 1200 K. An ab initio-based pairwise model is developed to describe the interactions that take place at the Ni – FLiNaK interface. It was shown that at temperatures of 800 K and 1000 K, lithium and fluorine ions are predominantly adsorbed on the nickel surface in the form of a two-dimensional ordered structure. Such a dense layer prevents the dissolution of nickel and the metal solid has no defects in the surface layer. However, at a temperature of 1200 K the structure of the adsorption layer is noticeably disturbed with the partial replacement of lithium ions by sodium ones. Along with higher temperature, this leads to the formation of point defects and degradation of the surface layers of the nickel crystal lattice.
Keywords
Full Text:
PDFReferences
Olson L, Sridharan K, Anderson M, Allen T, Nickel-plating for active metal dissolution resistance in molten fluoride salts, J. Nucl. Mater., 411 (2011) 51–59. https://doi.org/10.1016/j.jnucmat.2011.01.032
Wang Y, Zhang S, Ji X, Wang P, et al., Material corrosion in molten fluoride salts, Int. J. Electrochem. Sci., 13 (2018) 4891–4900. https://doi.org/10.1016/10.20964/2018.05.33
DorMahammadi H, Pang Q, Murkute P, Arnadottir L, et al., Investigation of chloride-induced depassivation of iron in alkaline media by reactive force field molecular dynamics, Materials Degradation, 3 (2019) 19. https://doi.org/10.1038/s41529-019-0081-6
Dong C, Ji Y, Wei X, Xu A, et al., Integrated computation of corrosion: Modelling, simulation and applications, Corrosion Communications, 2 (2021) 8–23. https://doi.org/10.1016/j.corcom.2021.07.001
Rudenko A, Redkin A, Il’ina E, Pershina S, et al., Thermal conductivity of FLiNaK in a molten state, Materials, 15 (2022) 5603. https://doi.org/10.3390/ma15165603
Ghods P, Isgor OB, Brown JR, Bensebaa F, et al., XPS depth profiling study on the passive oxide film of carbon steel in saturated calcium hydroxide solution and the effect of chloride on the film properties, Appl. Surface Sci., 257 (2011) 4669–4677. https://doi.org/10.1016/j.apsusc.2010.12.120
Zakiryanov D, Kobelev M, Tkachev N, Melting properties of alkali halides and the cation-anion size difference: A molecular dynamics study, Fluid Phase Equil., 506 (2020) 112369. https://doi.org/10.1016/j.fluid.2019.112369
Zakiryanov DO, Applying the Born-Mayer model to describe the physicochemical properties of FLiNaK ternary melt, Comp. Theor. Chem., 1219 (2023) 113951. https://doi.org/10.1016/j.comptc.2022.113951
Foiles SM, Baskes MI, Daw MS, Embedded-atom-method functions for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, and their alloys, Phys. Rev. B, 33 (1986) 7983–7991. https://doi.org/10.1103/PhysRevB.33.7983
Morse PM, Diatomic molecules according to the wave mechanics. II. Vibrational levels, Phys. Rev., 34 (1929) 57–64. https://doi.org/10.1103/PhysRev.34.57
Latorre CA, Ewen JP, Gattinoni C, Dini D, Simulating surfactant – iron oxide interfaces: From density functional theory to molecular dynamics, J. Phys. Chem. B, 123 (2019) 6870–6881. https://doi.org/10.1021/acs.jpcb.9b02925
Guymon CG, Rowley RL, Harb JN, Wheeler DR, Simulating an electrochemical interface using charge dynamics, Cond. Matter Phys., 8(2) (2005) 335–356. https://doi.org/10.5488/CMP.8.2.335
Adamo C, Barone V, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 110 (1999) 6158–6170. https://doi.org/10.1063/1.478522
Grimme S, Antony J, Ehrlich S, Krieg H, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 132 (2010) 154104. https://doi.org/10.1063/1.3382344
Weigend F, Accurate coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 8 (2006) 1057–1065. https://doi.org/10.1039/B515623H
Nosé S, Constant temperature molecular dynamics methods, Prog. Theor. Phys. Supp., 103 (1991) 1–46. https://doi.org/10.1143/PTPS.103.1
Thompson HM, Aktulga R, Berger DC, Bolintineanu WM, et al., LAMMPS – a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Comp. Phys. Comm., 271 (2022) 10817. https://doi.org/10.1016/j.cpc.2021.108171
Fumi FG, Tosi MP, Ionic sizes and born repulsive parameters in the NaCl-type alkali halides – I The Huggins-Mayer and Pauling forms, J. Phys. Chem. Solids., 25 (1964) 31–43. https://doi.org/10.1016/0022-3697(64)90159-3
Cahn R, Crystal defects and melting, Nature, 273 (1978) 491–492. https://doi.org/10.1038/273491b0
DOI: https://doi.org/10.15826/elmattech.2023.2.023
Copyright (c) 2023 Dmitry O. Zakiryanov, Mikhail A. Kobelev

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
![]()
![]()
![]()
![]()